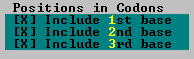Alt + M
-
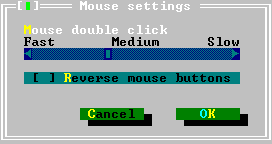
* how fast a double-click is, and
* which mouse button (right or left) is active.
Alt + F1
In this chapter, all the menus and commands that are available in the user-interface will be discussed. Many of these commands are tied to the hot-keys displayed next to them. These hot-keys sometimes require two or more keys. Such combinations are shown with a + sign to indicate that these keys should be pressed simultaneously.
| MEGA menu Alt + M |
||
| About MEGA | It opens a window with information about the current version of MEGA copyright notice, and the name and addresses of the authors for correspondence. | |
| Reference | It gives a way of citing MEGA in the "Literature Cited" section of research articles. | |
| Mouse | The Mouse command brings up a dialog box for selection of various mouse control options, including:
| |
| Calculator | It opens a simple four function calculator that can be operated with the keyboard as well as the mouse. | |
| Calendar | The Calendar displays the current month, highlighting today's date. The next and previous months are viewed with the '+' and '-' keys. Clicking the mouse on triangle icons also changes the month. | |
| Thank You | A Thank You note for help in development of MEGA is included here. | |
| Using Help Alt + F1 | Help on the on-line context-sensitive help is provided here. | |
| File menu Alt + F | |||
| Browse F5 |
The Browse command brings up a File Name box where the file to be opened is specified. This file is displayed on the screen in the read-only mode. Use the cursor movement keys to move around in the file or use the mouse on the scroll bars (see chapter 6 for details). | ||
| Edit | This editor supports most of the basic editing functions, including: saving file, manipulating text-blocks, and finding and replacing text-strings (see chapter 6 for more details). | ||
| Open File F3 | It displays a File Name dialog box for selecting the file to edit. The file is opened for editing in a new window. Many files can be opened for editing simultaneously in this editor. | ||
| Create New File | This command creates a new file with a temporary file name UNTITLED for editing. MEGA will require a file name whenever the file is saved. | ||
| Save File F2 | The currently edited file is saved on the disk with this command. If an UNTITLED file is saved, then a dialog box will prompt for the file name to store the text. | ||
| Save As Alt+F2 | A File Nane box prompts for a name to save the current file with a new file name. | ||
| Cut Shift + Del | Cut removes the selected text from the current file and places it in the clipboard, A Paste operation will retrieve this text when desired. Text can be pasted many times and to many files. | ||
| Copy Ctrl + Ins | Copy keeps the selected text intact and places its copy in the clipboard for the Paste command that can retrieve this text in any desired editor window. | ||
| Paste Shift + Ins | With this command, the most recently selected text in the clipboard is inserted into the current file at the cursor position. | ||
| Clear Ctrl + Del | Removes the selected text from the current document. The cleared text is not retrievable. | ||
| Undo Ctrl + U | Undo reverses the last editing command and restores the text. It works only on the last modified line. | ||
| Find String Ctrl + Q F | This command displays the Find Text dialog box in which the text to be searched is entered. | ||
| Replace String Ctrl + Q A | It displays the Find and Replace box where the text-string to be searched and replaced is entered along with the replacement string. | ||
| Show Clipboard | Shows the contents of the clipboard and the currently selected text block for Paste operations. | ||
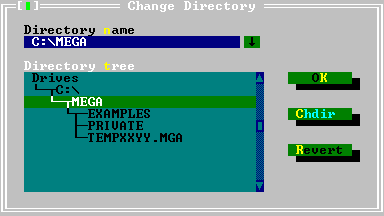
| Change Dir | |||
| The Change Directory dialog box consists of a directory input box, directory tree list box, and three buttons: OK, Chdir, and Revert.
The Chdir button changes the current directory once you select or type in a directory name. The Revert button lets you go back to the previous directory, as long as you have not exited the dialog box. The Directory Tree list box provides navigation through the directory structure with selection bar and Alt + C (short cut for Chdir command). If you are using the keyboard, press Enter to choose the changes made. Press Esc to cancel the changes made. | |||
| DOS Shell | With the DOS Shell command, you can leave the program temporarily to perform DOS commands and run other programs. To return to MEGA, type EXIT at the DOS prompt. | ||
| Exit MEGA Alt + X | This command terminates the current session of MEGA. It deletes all options and cleans all the temporary files from the disk. | ||
| Data menu Alt + D | |||
| Activation of the data file is the first step in data analysis. As soon as a data set becomes active, relevant in-memory data editing options are enabled depending on the input data. Virtually any subset of the original data can be selected through options for selecting OTUs and sites (or codons). A detailed description of in-memory editing options is given in chapter 2. Selected subset data can be examined with the Data Presentation command. | |||
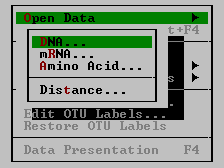
| Open Data | This command produces a submenu with four options. This command is enabled only if no other data set is active.
| ||
| Close Data Alt + F4 | It inactivates the current data set. Before doing so, it reconfirms your action. To save in-memory data editing, use Export command from the Data Presentation option. | ||
| Select OTUs | The OTUs scan be deleted (or re-inserted) with this option obviating any need for the modification of original input data file. The Select OTUs command brings up a list of OTU labels. In this list, some labels are checked ( Ö ) and others are not. Presence of a Ö mark indicates that the OTU is included in the current data subset. You can delete or insert an OTU by using the Del or Ins keys. To have the changes made, press Enter (click on OK) or abort the current changes with the Esc key (clicking on Cancel). | ||
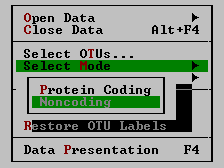
| Select Mode | This command is available for DNA and mRNA sequences only. It brings up a submenu where protein-coding or non-coding mode of analysis can be chosen.
| ||
| Select Sites/Codons | Desired sites/codons can be selected with this option.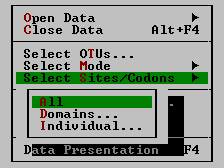
| ||
| All | All inserts all the sites (or codons depending on the mode) into the data subset for analysis. It is selected by default whenever a new data file is opened. | ||
| Domains | With this command a subset of original data containing up to 10 disjoint domains can be selected. This option can be useful if multiple exons exist for the gene studied.
The Domain command queries about the number of domains in the sequence first. You may enter any value from I to 10 (for more than 10 domains use Individual option). The location of domains in terms of their position in the input sequences is then entered in the next dialog box. All the domains must be non-overlapping and should be specified in the order from left to right. | ||
| Individual | This option is useful if the sites (or codons) to be analyzed are spread over the length of the sequence. Selection of this option produces a list of numbers corresponding to sites (or codons). Check ( Ö ) mark before a site (or codon) number indicates its inclusion in the data subset. Use the Ins and Del keys to insert and delete the desired sites and codons. The changes made can be accepted with the Enter key and discarded with the Esc key. | ||
| Select Outgroups | Outgroups are selected with this option. None of the methods in MEGA is sensitive to outgroups, but they are used when displaying phylogenetic trees on the screen. A list box containing all current OTU labels is displayed on selection of this option. In this list, checked ( Ö ) labels are designated as outgroups. Use the Ins and Del keys to include and exclude OTUs from the set of outgroups. To have the changes made, press Enter or click on OK; to abort, press Esc key or click on Cancel. | ||
| Edit OTU Labels | Often OTU labels present in the input file are required to be altered for publication purposes. This can be done in MEGA without affecting the input data file by using the Edit OTU Labels command. This command displays a list box dialog that contains a list of all the OTU labels. To edit an OTU label: press Space bar (or double click) on the OTU label and modify the existing label in the input line box produced, and press Enter to make the change. Labels of many OTUs can be edited at the same time. All the changes made can be accepted by pressing Enter finally or discarded by pressing Esc. MEGA does not require unique OTU labels, but you may find it difficult to identify OTUs individually. | ||
| Restore OTU Labels | OTUs labels are converted back to their original names as specified in the input data file. | ||
| Data Presentation F4 | This module provides an assortment of useful functions. It contains commands for exporting current data subsets to files in various formats; highlighting variable, parsimony-informative, and two- and fourfold redundant sites; translating nucleotide sequences; and computing sequence statistics such as base compositions, codon usage, alignment gap frequencies, and the variability in sliding windows. Selection of Data|Display Sequence Data command displays "Current Data" window that is described in detail in chapter 6. | ||
| Distance menu Alt + T | ||
| Commands on this menu are used to select a distance measure from three submenus: Nucleotide, Syn-Nonsynonymous, and Amino Acid. Distances are computed and saved to files with Compute Distances command. | ||
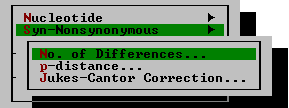
| Nucleotide | This command is available only for nucleotide sequences. It presents a choice among seven distance measures. Selection of a distance measure does not automatically initiate the distance computation; it only selects the distance measure to be used. The following submenu appears on selection of the Nucleotide command:
| |
| Syn-Nonsynonymous | This command is available for protein-coding nucleotide sequences only. It brings up a submenu containing three methods. After selecting a distance measure, you will be required to specify whether the synonymous or the nonsynonymous distances are to be computed. (See discussion in chapter 4.)
| |
| Amino Acid | This command is enabled for amino acid sequences and protein-coding nucleotide sequences. Nucleotide sequences are automatically translated into amino acid sequences by using the selected genetic code table.
| |
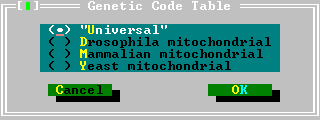
| Genetic Code Table | This command produces a dialog box with four radio-buttons. A genetic code table can be selected by double clicking (or by using arrow keys) on the desired code table. The changes made are discarded by pressing Esc (or by clicking on Cancel); and accepted by pressing Enter (or clicking on OK).
| |
| Compute Distances F7 | This command is for estimating pairwise distances. It should only be used if you want the distances are to be written into a text file. It is not necessary to compute distances using this command for estimating phylogenetic trees, because the distances are obtained automatically by the Construct Tree(s) command.
Selection of this command brings up a dialog box with options such as precision, page width, codon positions, etc.
| |
| Phylogeny menu Alt + P | ||
| This menu contains options for selecting tree-building methods and conducting test of their interior branch lengths. | ||
| UPGMA | This command is used to select the Unweighted Pair Group Method with Arithmetic means (UPGMA) for phylogenetic reconstruction (see chapter 5 for details.) | |
| Neighbor-Joining | The neighbor-joining (NJ) method is selected with this command (see chapter 5 for details). | |
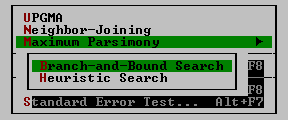
| Maximum Parsimony | In this method you have two options: the branch-and-bound search and the heuristic search (see chapter 5 for details).
| |
| Construct Tree(s) F8 | This command initiates the reconstruction of a phylogeny by using the selected tree-building method and the distance measure (if applicable). It automatically computes the distances, if required, and presents the tree on the screen in the phylogenetic-tree editor (see chapter 6).
| |
| Bootstrap Test Alt + F8 | The bootstrap test is used for assessing the reliability of a tree obtained (see chapter 5). MEGA provides a bootstrap test for UPGMA and the NJ method only. In response to the Bootstrap Test command, a dialog box appears with various options. Many options in this dialog box are similar to the ones described above for the Construct Tree(s) command. Therefore, only the options that are not described before are discussed below.
| |
| Standard Error Test Alt + F7 | This command is to conduct the standard error test for interior branch Iengths of a neighbor-joining tree. This option is available only for nucleotide sequence data whenever p-distance, Jukes-Cantor distance, or Kimura's 2-parameter distance is used (see chapter 5).
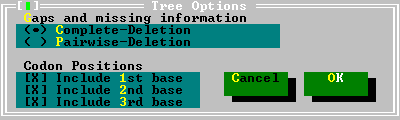
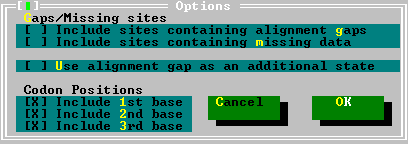
In this test, MEGA removes all sites containing alignment gaps and missing-information sites (Complete-Deletion option) from the sequence data. If the nucleotide sequence data is used in the coding mode, the choice of codon positions is available. | |
| Window menu Alt + W | ||
| The Window menu contains commands to close, move, and perform other window-management commands. Most of the windows in this program have all the standard window elements, including scroll bars, a close box, and zoom icons. | ||
| Resize/Move Alt + F9 | Choose this command to change the size and position of the active window. | |
| Size | If you press Shift with an arrow key, the size of the active window will be altered. Once you've adjusted its size or position, press Enter. If a window has a re-size corner, you can drag that corner to resize the window. | |
| Move | When the Window|Re-size/Move is chosen, the active window moves in response to the arrow keys. Press Enter after the window has been moved. With the mouse, a window is moved by dragging its title bar with the mouse. | |
| Zoom F9 | Choose Zoom to resize the active window to the maximum size. If a window is already zoomed, this command restores it to its previous size. You may also double-click on the zoom-icon on the window's title bar to zoom or un-zoom. | |
| Tile | The Title command arranges all the windows on the screen in the following manner:
| |
| Cascade | The Window|Cascade command stacks all the windows on desktop as shown below.
| |
| Next F6 | Choose Next to cycle forwards through the windows on the desktop. | |
| Previous Alt + F6 | Previous cycles backwards through the windows on the desktop. | |
| Close | Choose Close to remove the active window. The Close window icon on Alt+F3 the upper right corner can also be used to close any window. | |